Rhabdomyosarcoma Causes Symptoms and Treatment Options
As of late 2023, Rhabdomyosarcoma (RMS) remains the most common type of soft tissue sarcoma found in children and adolescents, though it can also occur in adults. This aggressive cancer arises from mesenchymal cells that have failed to fully differentiate into skeletal muscle cells. Despite skeletal muscle not being present in certain parts of the body, such as the bladder or the middle ear, RMS can develop virtually anywhere these precursor cells exist. Due to its rarity and complexity, RMS requires immediate attention from specialized medical centers utilizing multidisciplinary oncology teams. Understanding the pathology, symptoms, and modern treatment modalities is crucial for early detection and improved outcomes. This article provides a comprehensive, fact-based overview of the current medical understanding of Rhabdomyosarcoma, intended for informational purposes to assist patients, families, and healthcare seekers in navigating this diagnosis.

What is Rhabdomyosarcoma?
Rhabdomyosarcoma is a malignant neoplasm characterized by the proliferation of cells that resemble rhabdomyoblasts—immature muscle cells. It accounts for approximately 3% of all childhood cancers and about 40% of all soft tissue sarcomas in children. The disease is fundamentally different from adult soft tissue sarcomas in terms of biology and responsiveness to therapy. The primary challenge in treating RMS lies in its propensity for local invasion and early metastasis, often spreading to the lungs, bone marrow, and bones.
Medical classification separates RMS into distinct histological subtypes, which dictate the aggressiveness of treatment and the overall prognosis. The two most common subtypes are Embryonal Rhabdomyosarcoma (ERMS) and Alveolar Rhabdomyosarcoma (ARMS). While ERMS is more common and generally has a favorable prognosis, ARMS is associated with a higher risk of metastasis and recurrence.
Major Histological Subtypes of Rhabdomyosarcoma
Identifying the specific subtype of Rhabdomyosarcoma is a critical step in the diagnostic process. Pathologists utilize immunohistochemistry and cytogenetic analysis to distinguish between these variants.
H2: Primary Histological Subtypes
-
Embryonal Rhabdomyosarcoma (ERMS):
- Demographics: Most common in children under 10 years old.
- Locations: Frequently found in the head and neck region (orbit, nasopharynx) or the genitourinary tract.
- Genetics: Often associated with loss of heterozygosity at chromosome 11p15.5.
- Prognosis: Generally favorable with standard multimodal therapy.
-
Alveolar Rhabdomyosarcoma (ARMS):
- Demographics: More common in adolescents and young adults.
- Locations: Predominantly affects the trunk, extremities, and perineal/para-testicular regions.
- Genetics: Characterized by specific chromosomal translocations, primarily t(2;13)(q35;q14).
- Prognosis: Aggressive; higher risk of treatment failure compared to ERMS.
Rare and Pleomorphic Subtypes
- Botryoid Rhabdomyosarcoma: A variant of ERMS that presents as a "grape-like" mass, usually arising in hollow organs such as the vagina or bladder in young children.
- Spindle Cell/Sclerosing Rhabdomyosarcoma: Can occur in children (paratesticular region) or adults; tends to have an indolent course in children but is aggressive in adults.
- Pleomorphic Rhabdomyosarcoma: Typically found in adults; involves cells that look wildly disorganized under a microscope; poor prognosis.
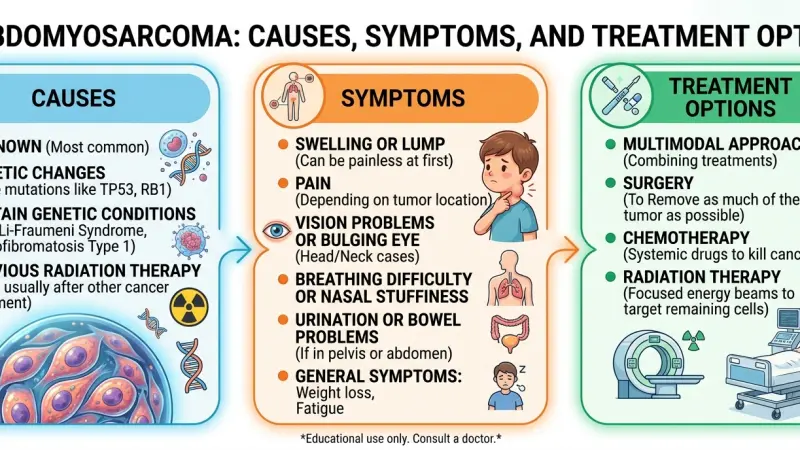
Causes and Risk Factors
The etiology of Rhabdomyosarcoma involves a complex interplay of genetic mutations and developmental biology. However, for the majority of patients, no clear environmental cause or lifestyle factor is identifiable.
Genetic Predispositions and Syndromes
- Li-Fraumeni Syndrome: A hereditary disorder caused by mutations in the TP53 tumor suppressor gene, significantly increasing the risk of various cancers, including RMS.
- Beckwith-Wiedemann Syndrome: An overgrowth syndrome linked to chromosome 11p15.5 abnormalities.
- Neurofibromatosis Type 1 (NF1): A condition causing tumors to grow on nerves; patients have a slightly elevated risk of RMS.
- Costello Syndrome: A rare disorder caused by HRAS mutations.
- Noonan Syndrome: A genetic disorder that prevents normal development in various parts of the body.
Environmental and Biological Factors
- Parental Age: Some studies suggest a correlation between advanced parental age and the incidence of RMS in offspring, though this link remains under investigation.
- In Utero Exposure: Limited data suggests potential associations with X-ray exposure during pregnancy, but evidence is not conclusive.
- Random Mutations: In most cases, the genetic changes causing RMS are somatic, meaning they occur randomly during cell division in the child's development and are not inherited.
Common Signs and Symptoms
The clinical presentation of Rhabdomyosarcoma varies significantly depending on the tumor location, size, and extent of invasion. Because symptoms often mimic common childhood injuries or infections, diagnosis can be delayed.
Symptoms by Anatomical Location
- Head and Neck (40% of cases):
- Orbital RMS: Unilateral bulging of the eye (proptosis), vision changes, drooping eyelid, sinus congestion.
- Parameningeal RMS: Headaches, facial pain, cranial nerve palsies, nasal discharge, ear discharge (often bloody), or difficulty swallowing.
- Genitourinary Tract (20% of cases):
- Bladder/Prostate: Difficulty urinating (urinary retention), blood in urine (hematuria), or a palpable abdominal mass.
- Vaginal: Vaginal bleeding or a protruding mass in infants and young girls.
- Paratesticular: Painless, firm mass in the scrotum or groin; may be mistaken for a hydrocele.
- Extremities (30% of cases):
- Arms and Legs: Painless or tender lump/swelling in the muscle; rapid growth of the mass.
- Trunk/Retroperitoneum:
- Abdominal distension, back pain, or constipation due to mass effect.
Diagnostic Procedures and Staging
A definitive diagnosis of Rhabdomyosarcoma requires a series of rigorous tests. The goal is not only to confirm the presence of cancer cells but also to determine the extent of the disease (staging).
Key Diagnostic Steps
- Biopsy: The gold standard for diagnosis.
- Incisional Biopsy: Surgical removal of a small piece of the tumor.
- Needle Biopsy: Less invasive; uses a hollow needle to extract tissue.
- Pathology: Tissue samples are stained for muscle-specific proteins like Myogenin, MyoD1, and Desmin.
- Imaging Studies:
- MRI: Provides detailed images of soft tissue involvement; crucial for head/neck and extremity tumors.
- CT Scan: Used to assess the chest for metastasis and abdomen/pelvis for lymph node involvement.
- PET Scan: Metabolic imaging to detect active cancer cells throughout the body.
- Bone Scan: Detects spread to the skeletal system.
- Laboratory Tests:
- Bone Marrow Aspiration and Biopsy: Checks for cancer cells in the bone marrow (essential for high-stage RMS).
- CBC (Complete Blood Count): Assesses general health and marrow function.
Staging and Risk Grouping Medical professionals utilize the TNM Staging System (Tumor, Nodes, Metastasis) and the Clinical Grouping System (based on surgical resection) to classify the disease.
- Clinical Group I: Disease is completely resected with negative margins.
- Clinical Group II: Microscopic residual disease remains after surgery.
- Clinical Group III: Gross residual disease remains after surgery/biopsy.
- Clinical Group IV: Distant metastatic disease is present at diagnosis.
Treatment Protocols and Modalities
Treatment for Rhabdomyosarcoma is multimodal, involving a combination of chemotherapy, surgery, and radiation therapy. The specific regimen is determined by the risk group (Low, Intermediate, or High), which considers the site, size, resectability, and histology.
Chemotherapy
- Induction Phase: Shrinks the tumor to make surgery or radiation more effective.
- Maintenance Phase: Kills remaining cancer cells to prevent recurrence.
- Common Agents:
- Vincristine: A vinca alkaloid that inhibits cell division.
- Actinomycin D: An antibiotic that interferes with RNA transcription.
- Cyclophosphamide/Ifosfamide: Alkylating agents that damage DNA.
- The standard combination is known as VAC (Vincristine, Actinomycin, Cyclophosphamide).
Radiation Therapy
- Indications: Used when surgical removal would result in significant disfigurement or functional loss, or when residual disease is present after surgery (Groups II and III).
- Types:
- External Beam Radiation Therapy (EBRT): The most common method.
- Intensity-Modulated Radiation Therapy (IMRT): Shapes the radiation beams to match the tumor, sparing healthy tissue.
- Proton Therapy: Precise delivery of radiation, highly recommended for pediatric patients to reduce long-term side effects.
Surgery
- Goal: Complete resection of the primary tumor with a margin of healthy tissue.
- Conservative Approaches: Surgeons prioritize limb-sparing procedures and organ preservation.
- Second-Look Surgery: Performed after initial chemotherapy to remove residual mass if possible.
Prognosis and Survival Rates
The prognosis for Rhabdomyosarcoma has improved significantly over the last few decades due to collaborative research trials (such as those by the Children’s Oncology Group). Survival rates are statistical estimates and cannot predict individual outcomes.
Survival Data Based on Risk Groups
- Low Risk:
- Favorable Sites: Orbit, bladder/prostate (biliary tract), or head/neck (parameningeal) with completely resected disease.
- 5-Year Survival: Generally exceeds 70% to 90%.
- Intermediate Risk:
- Unfavorable Sites: Bladder/prostate (non-biliary), extremities, or cranial parameningeal tumors.
- Criteria: Tumors >5cm or with lymph node involvement.
- 5-Year Survival: Approximately 50% to 70%.
- High Risk:
- Metastatic Disease: Tumors that have spread to distant sites like lungs, bone, or bone marrow at diagnosis.
- 5-Year Survival: Historically lower than 30%, though clinical trials continue to improve these figures.
Featured Snippet Optimization
Rhabdomyosarcoma (RMS) is a rare cancer that forms in soft tissue, specifically cells that develop into skeletal muscles. It is most common in children but can occur in adults. Symptoms vary by location but often include a painless lump or swelling. Standard treatment involves a combination of chemotherapy, surgery, and radiation therapy.
Long-Term Follow-Up and Survivorship
Survivors of Rhabdomyosarcoma require lifelong monitoring due to the potential for late effects caused by aggressive treatments. These late effects can manifest years after the completion of therapy.
Common Long-Term Health Considerations
- Cardiac Toxicity: Caused by chemotherapy agents like anthracyclines (though less common in RMS protocols than other cancers) and radiation to the chest.
- Musculoskeletal Issues: Growth abnormalities in irradiated areas; limb length discrepancies.
- Infertility: Potential side effect of high-dose alkylating agents (Cyclophosphamide).
- Secondary Malignancies: Increased risk of developing other cancers later in life due to radiation and chemotherapy exposure.
- Hearing Loss: Ototoxicity from platinum-based chemotherapies if used.
Follow-up care typically involves annual physical exams, imaging of the primary site, and monitoring of organ function (heart, lungs, kidneys). Psychological support is also essential to address the social and emotional challenges faced by childhood cancer survivors.
Rhabdomyosarcoma
Rhabdomyosarcoma represents a complex and challenging category of pediatric and adult oncology. The differentiation between Embryonal and Alveolar subtypes, combined with precise staging and risk grouping, dictates the therapeutic pathway. Modern medicine utilizes a three-pronged attack strategy involving systemic chemotherapy, local radiation, and surgical excision. While survival rates for localized disease are high, metastatic RMS remains a critical area of ongoing research. The medical community continues to refine protocols to maximize efficacy while minimizing the long-term toxicities associated with current standard treatments. For patients and families, understanding the biological nature of this sarcoma is the first step in managing the rigorous treatment journey ahead.
FAQs
1. Is Rhabdomyosarcoma considered curable? Yes, localized Rhabdomyosarcoma is often curable. Approximately 70% of children with localized RMS survive long-term. Cure rates are lower if the cancer has spread to distant parts of the body.
2. Who is most at risk for Rhabdomyosarcoma? It is primarily a disease of childhood, with the majority of cases diagnosed in children under the age of 10. However, it can also occur in adolescents and adults, often with a more aggressive presentation.
3. What is the most common location for Rhabdomyosarcoma? The head and neck region is the most common site, particularly around
the eye (orbit) and in the nasopharynx. The genitourinary tract and extremities are also frequent locations.
4. Is Rhabdomyosarcoma hereditary? Most cases are sporadic, meaning they occur by chance. However, certain inherited genetic syndromes, such as Li-Fraumeni syndrome and Beckwith-Wiedemann syndrome, increase the risk.
5. How is Rhabdomyosarcoma diagnosed? Diagnosis is confirmed via a biopsy of the tumor. Doctors also use imaging studies like MRI, CT scans, and bone marrow biopsies to determine the stage of the disease.
6. Does Rhabdomyosarcoma hurt? Some tumors present as painless lumps, particularly in the extremities. However, others can cause pain if they compress nerves or organs, or if they invade bone.
7. What is the VAC regimen in Rhabdomyosarcoma treatment? VAC stands for the combination of three chemotherapy drugs: Vincristine, Actinomycin D, and Cyclophosphamide. It is a standard backbone for treating intermediate and high-risk RMS.
8. What are the side effects of Rhabdomyosarcoma treatment? Side effects include nausea, hair loss, increased risk of infection, fatigue, and anemia. Long-term side effects can involve growth issues, infertility, and heart problems.
9. Can Rhabdomyosarcoma come back? Yes, RMS can recur. Relapse typically happens within the first 3 years after treatment but can occur later. Recurrent RMS is more difficult to treat and often requires different clinical trials.
10. Is surgery always required for Rhabdomyosarcoma? Not always. If the tumor is in a location where surgery would remove vital organs or cause disfigurement (e.g., behind the eye), doctors may rely on chemotherapy and radiation to shrink and kill the cancer.
11. How long does treatment last? Treatment for Rhabdomyosarcoma is intensive and usually lasts between 6 months and 1 year, depending on the risk group and response to therapy.
12. What is the difference between ERMS and ARMS?ERMS (Embryonal) is more common, has a better prognosis, and usually occurs in younger children. ARMS (Alveolar) is rarer, more aggressive, often occurs in teens, and has distinct genetic translocations.
13. Can adults get Rhabdomyosarcoma? Yes, adults can get RMS, though it accounts for less than 1% of adult cancers. It behaves aggressively in adults and is often treated using pediatric protocols.
14. What are "late effects" in Rhabdomyosarcoma survivors? Late effects are health problems that appear months
or years after treatment, such as secondary cancers, heart disease, hormonal deficiencies, and learning difficulties.
15. Are there clinical trials for Rhabdomyosarcoma? Yes. Clinical trials are essential for improving outcomes, especially for high-risk and recurrent RMS. Trials may test new immunotherapies, targeted drugs, or modified chemotherapy schedules.

Disclaimer: The information provided in this article is for general informational and research purposes only. Company details, features, services, and market positions may change over time. Readers are advised to visit official company websites and conduct independent research before making any business decisions or purchasing services.
Most Searchable Keywords

Recent Blogs
-

Explore Supermarkets in Chepstow and the Surrounding Wye Valley
-
Elite Guide to Taxi and Private Hire Services in Hereford
-
Explore Fuel and Roadside Convenience Across Beckington Today
-
Explore Abergavenny Tyre Services with Expert Local Fitters
-
Claim Your HMRC UK Tax Refund Before Application Deadlines





Categories
- Accountants (295)
- Advertising Agencies (559)
- Architects (149)
- Automobiles (376)
- Beauty (300)
- Carpenters (146)
- Cleaning Services (386)
- Dentists (189)
- Driving (61)
- Electricians (215)
- Energy (7)
- Event Organiser (683)
- Finance (597)
- Guide (3358)
- Health (2205)
- Information technology (206)
- Legal Services (352)
- Logistics (6)
- Maintenance (65)
- Manufacturing (35)

Sponsored Listings in Property Maintenance


Related Blogs

Explore Supermarkets in Chepstow and the Surr...
Read this insightful article "Explore Supermarkets in Chepstow and the Surrounding Wye Valley" to expand your knowledge!

Elite Guide to Taxi and Private Hire Services...
Read this insightful article "Elite Guide to Taxi and Private Hire Services in Hereford" to expand your knowledge!

Explore Fuel and Roadside Convenience Across...
Read this insightful article "Explore Fuel and Roadside Convenience Across Beckington Today" to expand your knowledge!
Questions & Answers – Find What
You Need, Instantly!
How can I update my business listing?
Is it free to manage my business listing?
How long does it take for my updates to reflect?
Why is it important to keep my listing updated?
